When we first started working on pediatric GIST more than five years ago, our initial observations suggested a homogenous clinical and genetic subset, with predilection for females, gastric location and wild-type genotype . Subsequent analysis of additional patients expanded this view, revealing that up to one third of cases may occur in males, whose tumors may arise in the small bowel and harbor activating KIT/PDGFRA mutations . In addition, with longer follow-up, it became clear that some pediatric patients eventually develop secondary neoplasms diagnostic for Carney’s triad, obscuring the distinction from the more common form of pediatric GIST. Furthermore, when applying genomic approaches, it became clear that GIST in young adults (defined as older than 18- year of age) is a heterogeneous disease, most of them resembling the adult counterpart, although rare cases of young females with multifocal gastric disease may be documented in this age group.
One additional level of complexity was brought by the overlapping attributes between pediatric and adult wild-type GIST. As demonstrated by us and others, the adult and pediatric wild-type tumors lack the typical genomic alterations, showing mainly a diploid phenotype (Figure 1).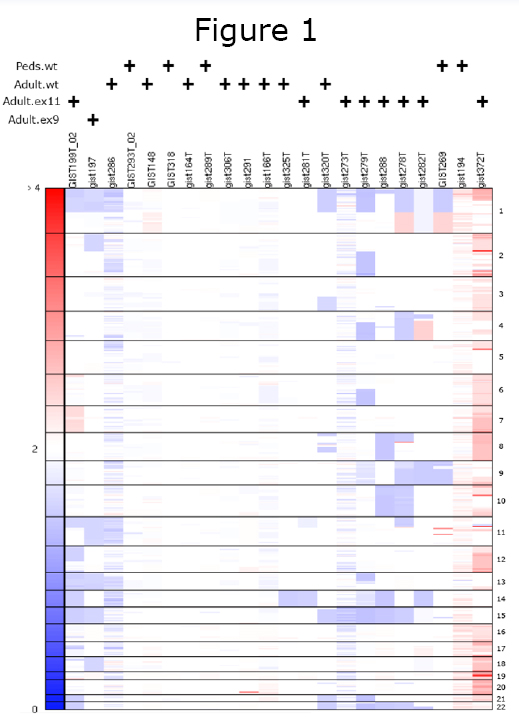 While loss of 14q and 22q occur frequently and non-randomly in both benign and malignant KIT mutated GIST, suggesting early molecular events, the absence of these genetic events in wild-type GIST suggest alternative pathways of tumor progression. Similar findings were also demonstrated by comparative genomic hybridization (CGH) technique used to analyze three Carney’s triad patients. The authors showed absence of the non-random 14q and 22q losses, characteristic of their sporadic counterpart, irrespective of site and biological grade. Furthermore, similar to wild type adult GIST patients, where clinical responses to imatinib are sparse and short-lived, pediatric GIST patients are consistently refractory to imatinib therapy. In vitro experiments concur with these clinical observations, showing an inferior degree of proliferation inhibition when treating Ba/F3 cells expressing wild type KIT with imatinib, as compared to nilotinib . For all the above reasons, at the joint initiative of the Life Raft Group and the National Insitutes of Health, a pediatric and wildtype adult GIST clinic was created to gather more insight on the similarities and difference between these two rare subsets. The initial response after the first three meetings was outstanding, the clinics being fully booked with patients from across the United States and Europe. Participating GIST clinicians and scientists were involved in a critical review of the clinical findings and management recommendations, as well as initiate a comprehensive tissue collection and data registry.
While loss of 14q and 22q occur frequently and non-randomly in both benign and malignant KIT mutated GIST, suggesting early molecular events, the absence of these genetic events in wild-type GIST suggest alternative pathways of tumor progression. Similar findings were also demonstrated by comparative genomic hybridization (CGH) technique used to analyze three Carney’s triad patients. The authors showed absence of the non-random 14q and 22q losses, characteristic of their sporadic counterpart, irrespective of site and biological grade. Furthermore, similar to wild type adult GIST patients, where clinical responses to imatinib are sparse and short-lived, pediatric GIST patients are consistently refractory to imatinib therapy. In vitro experiments concur with these clinical observations, showing an inferior degree of proliferation inhibition when treating Ba/F3 cells expressing wild type KIT with imatinib, as compared to nilotinib . For all the above reasons, at the joint initiative of the Life Raft Group and the National Insitutes of Health, a pediatric and wildtype adult GIST clinic was created to gather more insight on the similarities and difference between these two rare subsets. The initial response after the first three meetings was outstanding, the clinics being fully booked with patients from across the United States and Europe. Participating GIST clinicians and scientists were involved in a critical review of the clinical findings and management recommendations, as well as initiate a comprehensive tissue collection and data registry.
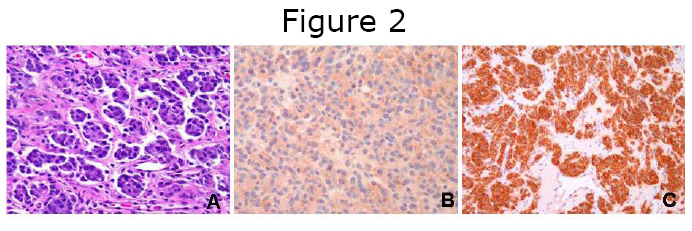
Because of their rarity, pediatric GISTs may pose significant challenges for the practicing surgical pathologist. Based on our experience, misdiagnoses may occur when limited biopsy material is available, composed of a proliferation of small undifferentiated cells. The differential diagnoses may then be restricted to entities considered in the family of small blue round cell tumors, such as Ewing sarcoma/Peripheral Neuroectodermal Tumor (PNET) or small-cell carcinoma. Neuroectodermal-type rosettes may be seen in pediatric as well as Carney’s triad GIST, and should not imply by itself a diagnosis of Ewing sarcoma/ PNET (Figure 2A). Furthermore, the immunohistochemical marker that is commonly expressed in Ewing sarcoma tumors (i.e. CD99), is hardly specific, being expressed in a variety of alternative diagnoses (Figure 2B). The diagnosis of primary Ewing sarcoma of the gastrointestinal tract is extraordinarily rare, and such diagnosis should be confirmed by molecular studies and identification of the EWS-ETS fusion transcript. It is our opinion that such challenging cases should be sent for expert pathology review, in large cancer centers with extensive experience in both pediatric and sarcoma pathology. After a correct diagnosis is rendered (diffuse and strong reactivity for CD117, Figure 2C), these patients should be referred for staging and management to sarcoma multidisciplinary teams in centers dedicated to pediatric GIST care.
If the pathologic criteria to define risk of malignancy in adult GIST are well established and confirmed by multiple studies (i.e. tumor size, mitotic activity), these factors are not reliable in predicting behavior in pediatric GIST. These patients frequently present with multiple nodules within the stomach, and thus the largest tumor dimension cannot be easily defined. Furthermore, a wide range in proliferation index can be detected between patients and even among multiple tumors from the same patient. Our experience is consistent with that of Miettinen, who noted that some pediatric patients with GIST developed metastasis despite being classified as low risk by criteria established in adult GIST. These findings suggest that GISTs in children are unpredictable, being more prone to metastasis than comparable gastric tumors in adults. Secondly, the biology of pediatric GIST appears to be more indolent than the adult counterpart, with long-term survival even in the presence of metastatic disease and without kinase inhibition therapy. Among the 12 pediatric patients with more than 6 months follow-up, all except two (83%) developed metastatic disease. In spite of such a high metastatic rate, only one patient has died so far, 138 months after initial diagnosis. Carney’s triad is a rare non-hereditary condition affecting young females, being characterized by two or more lesions, including GIST, pulmonary chondroma and extra-adrenal paraganglioma. The multifocal occurrence of the individual components of Carney’s triad suggests a genetic predisposition; however there is no evidence that the disorder is inherited. Less recognized, is the association of miscellaneous congenital malformations in a subset of Carney’s triad patients, which have not been described in GIST patients outside this syndromic setting. More recently it was recognized that the autosomal dominant inheritance of the dyad “paraganglioma and gastric GIST”, or “Carney-Stratakis syndrome” (CSS), represents a separate condition which affects both males and females and lacks the association with pulmonary chondromas. Mutations of the genes coding for succinate dehydrogenase subunits, typically associated with familial paragangliomas, are most likely implicated in the pathogenesis of CSS . As adult patients with wild-type GIST are among those achieving the most sustained clinical benefit on sunitinib therapy, the possibility of similar clinical efficacy in the pediatric age group remained in question. Recently, Janeway et al investigated this hypothesis on seven pediatric patients (aged 10-17 years) with advanced GIST treated with sunitinib on a treatment use protocol, after failing imatinib.
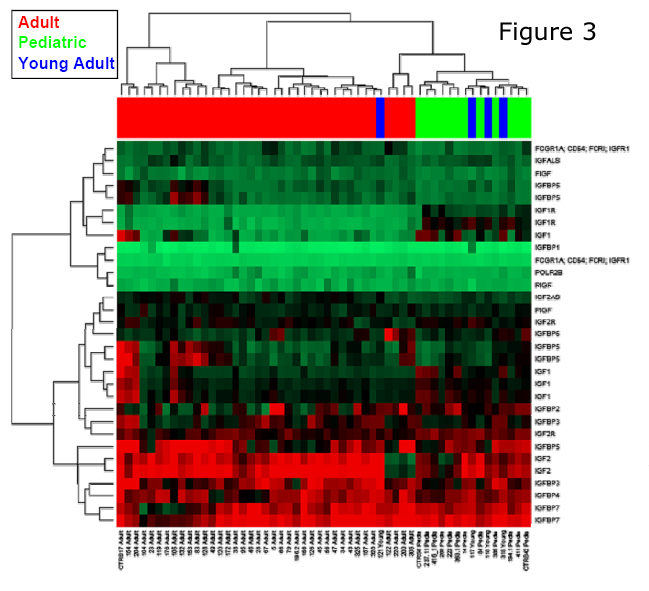
Their findings revealed a partial response in one patient, stable disease in five patients and disease progression in one patient. Thus six of seven patients showed benefit from sunitinib therapy for an average of 15 months, without significant toxicity. More recently, an interesting biomarker was depicted as a potential therapeutic target in pediatric GIST. Expression profiling identified the presence of IGF1R mRNA (Figure 3) overexpression in pediatric GIST, compared to adult GIST, including the wild-type cases. Additionally, Tarn and colleagues identified the presence of IGF1R amplification and protein overexpression in both wild-type and pediatric GIST 9. Applied immunoblotting and immunohistochemistry revealed that IGF1R was markedly overexpressed in the wild-type compared to mutant GIST. Although biochemical activation of the receptor was noted in all GIST tumors tested, regardless of KIT genotype, the levels of phospho-IGF1R did not correlate with the total IGF1R levels. However no activating IGF1R mutations were detected in the ten wild-type GIST tumors tested in any of the exons encoding juxtamembrane and kinases domains. Combination treatment experiments in GIST cell lines with NVP-AEW541 (an IGF1R inhibitor) and imatinib induced an additive strong cytotoxicity response. Follow-up studies are needed to confirm these initial results to support a solid scientific basis for future design of IGF1Rtargeted clinical trials. These results are highly relevant since a number of IGF1R antibodies are now available in clinical trials. Specifically, the SARC001 trial, using the IGF1R antibody from Roche Pharmaceuticals, was designed targeting pediatric and adult sarcomas, with specific emphasis on tumors characterized by recurrent chromosomal translocations. In keeping with these findings, the first pediatric GIST patient has been enrolled on an IGF1R-antibody protocol, after failing sunitinib therapy, with clinical response to be nvestigated.
References
- Prakash S, Sarran L, Socci N, et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol. 2005;27(4):179- 187.
- Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14(10):3204-3215.
- Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer research. 2007;67(19):9084-9088.
- Agaimy A, Pelz AF, Corless CL, et al. Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep. 2007;18(1):9-15.
- Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. The American journal of surgical pathology. 2005;29(10):1373-1381.
- Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108(2):132-139.
- Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79-88.
- Janeway KA, Albritton KH, Van Den Abbeele AD, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52(7):767-771.
- Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proceedings of the National Academy of Sciences of the United States of 2008;105(24):8387-8392.
Written by Cristina Antonescu, M.D.


